We present S-CGIB, a novel Architecture for Pre-training Graph Neural Networks in Molecular Structure Learning and developed by NS Lab, CUK based on pure PyTorch backend. The paper has been accepted for presentation at the AAAI 2025 conference.
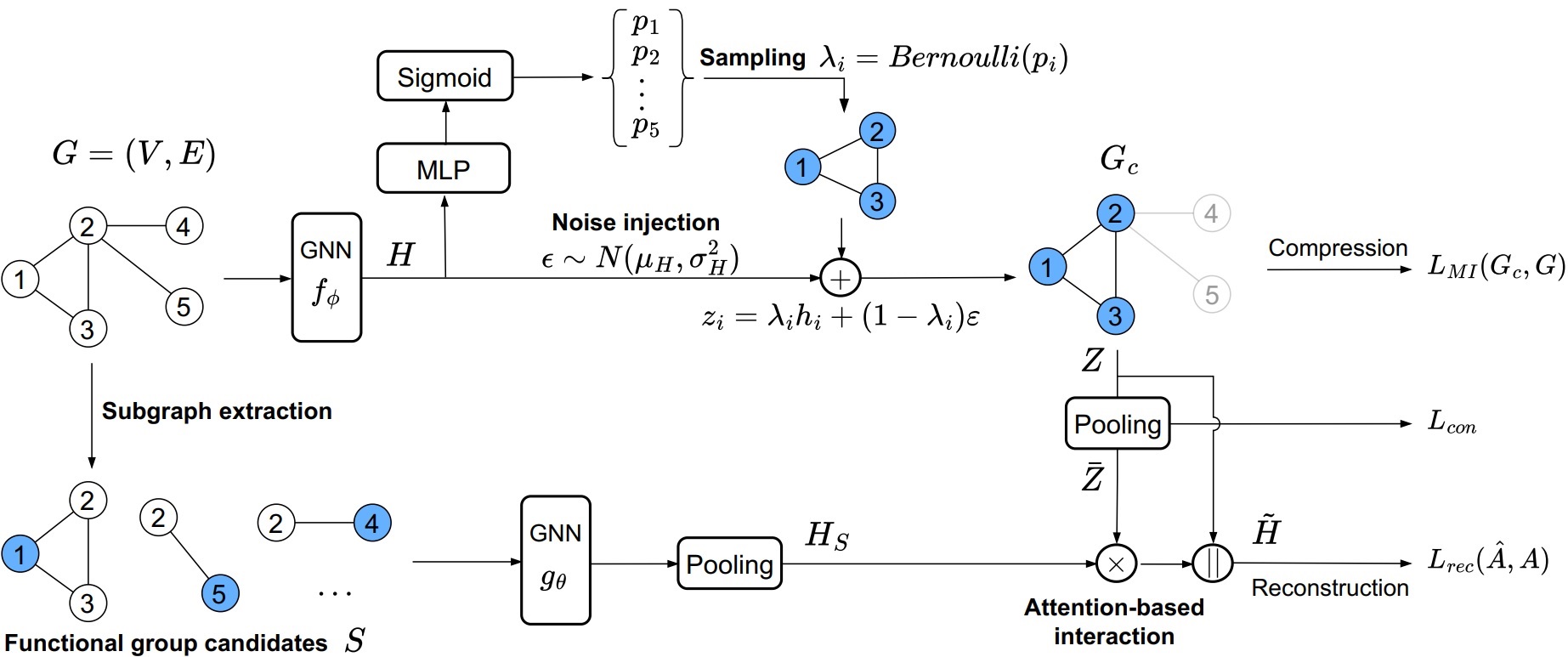
The overall architecture of Subgraph-conditioned Graph Information Bottleneck.
We aim to build a pre-trained Graph Neural Network (GNN) model on molecules without human annotations or prior knowledge. Although various attempts have been proposed to overcome limitations in acquiring labeled molecules, the previous pre-training methods still rely on semantic subgraphs, i.e., functional groups. Only focusing on the functional groups could overlook the graph-level distinctions. The key challenge to build a pre-trained GNN on molecules is how to (1) generate well-distinguished graph-level representations and (2) automatically discover the functional groups without prior knowledge. To solve it, we propose a novel Subgraph-conditioned Graph Information Bottleneck, named S-CGIB, for pre-training GNNs to recognize core subgraphs (graph cores) and significant subgraphs. The main idea is that the graph cores contain compressed and sufficient information that could generate well-distinguished graph-level representations and reconstruct the input graph conditioned on significant subgraphs across molecules under the S-CGIB principle. To discover significant subgraphs without prior knowledge about functional groups, we propose generating a set of functional group candidates, i.e., ego networks, and using an attention-based interaction between the graph core and the candidates. Despite being identified from self-supervised learning, our learned subgraphs match the real-world functional groups. Extensive experiments on molecule datasets across various domains demonstrate the superiority of S-CGIB.
A short description of S-CGIB:
- S-CGIB is trained to generate well-distinguished graph-level representations and automatically capture significant subgraphs without explicit annotations or prior knowledge in the context of Self-Supervised Learning. The fundamental idea behind our strategy is that, across the chemical domain, molecules share universal core subgraphs that can combine with specific significant subgraphs to robust representations of molecules.
- S-CGIB generates well-distinguished graph-level representations by compressing an input molecule graph into a graph core conditioned on specific significant subgraphs without using label information under the Subgraph-conditioned Graph Information Bottleneck principle.
- S-CGIB discover the significant subgraphs (functional groups) from the subgraph candidates (ego networks rooted at each node) through the attention-based interaction between the graph core and the ego networks under the graph reconstruction.
- Extensive experiments on molecule datasets across four domains demonstrate the superiority of S-CGIB.
The S-CGIB is available at:

Cite “S-CGIB” as:
Please cite our paper if you find S-CGIB useful in your work:
@InProceedings{Hoang_2025,
title={Pre-Training Graph Neural Networks on Molecules by Using Subgraph-Conditioned Graph Information Bottleneck},
volume={39},
url={https://ojs.aaai.org/index.php/AAAI/article/view/33891},
DOI={10.1609/aaai.v39i16.33891},
number={16},
booktitle = {Proceedings of the 39th AAAI Conference on Artificial Intelligence (AAAI 2025)},
author={Hoang, Van Thuy and Lee, O-Joun},
year={2025},
month={Apr.},
pages={17204-17213}
}
@misc{hoang2024pretraininggraphneuralnetworks,
title={Pre-training Graph Neural Networks on Molecules by Using Subgraph-Conditioned Graph Information Bottleneck},
author={Van Thuy Hoang and O-Joun Lee},
year={2024},
eprint={2412.15589},
archivePrefix={arXiv},
primaryClass={cs.LG},
url={https://arxiv.org/abs/2412.15589},
}
![]()
![]()
![]()
![]()
![]()
Contributors:
![]()
{kind=link}